
Since its launch in 2011 the ATB has grown dramatically driven by you the users.
We are now asking for your help in planning the next phase of the project.
The survey will only take 5 minutes and can be accessed here: ATB User Survey
Your feedback will help understand how:
Tautomers (Beta Test) for molecules in clinical phase 4 trials ≤ 30 atoms.
Display tautomers for e.g. Carbamazepine.
Search for more tautomers using the Existing Molecules tab.
*Please login to access this feature.
| Formula | C17H21N3O2 |
| Number of atoms | 43 |
| Net Charge | 0 |
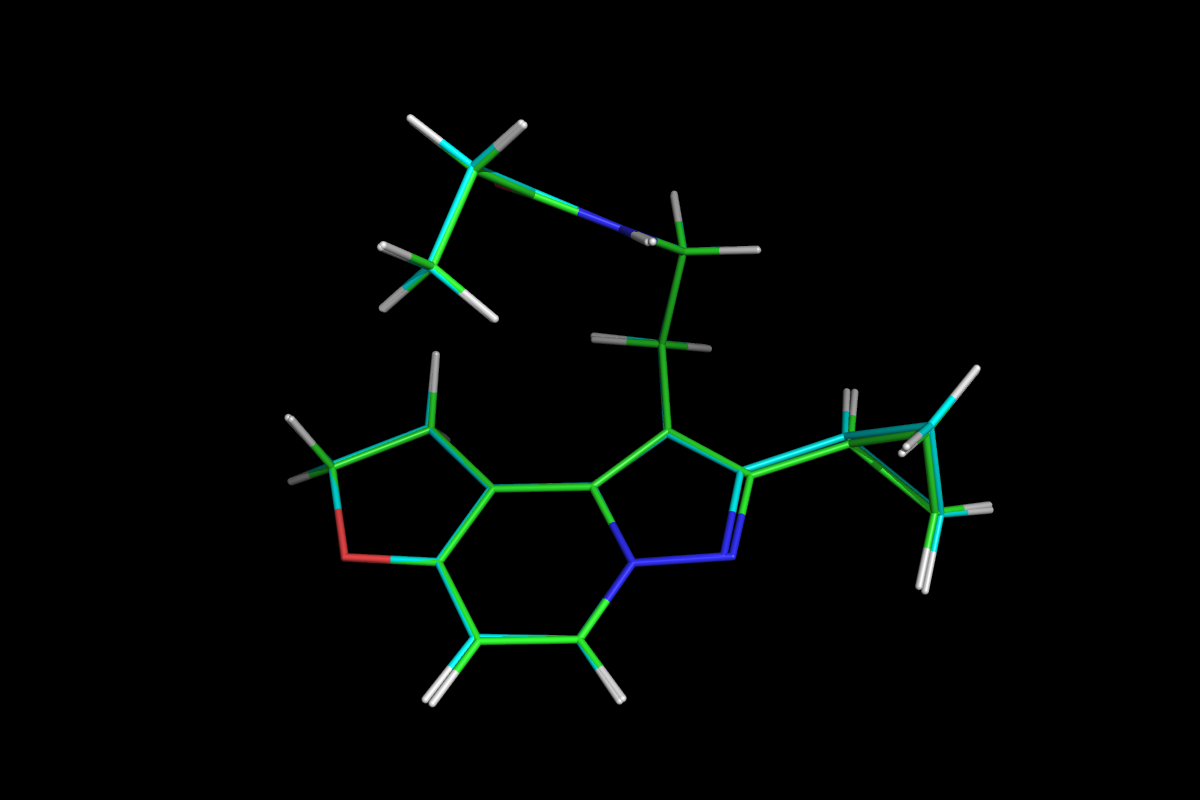
Superposition of QM optimised (green) and MM energy minimised (cyan) structures.
Click to toggle size.
| Pre-calculated molecules | 1,106,173 |
| Registered users | 25,160 |
| Downloads per month | 4,700 |
The ATB provides classical molecular force fields for novel compounds. Applications include:
This site provides:
Description
Malde AK, Zuo L, Breeze M, Stroet M, Poger D, Nair PC, Oostenbrink C, Mark AE.
An Automated force field Topology Builder (ATB) and repository: version 1.0.
J. Chem. Theory Comput., 2011, 7, 4026-4037. DOI:10.1021/ct200196m
Stroet M, Caron B, Engler MS, van der Woning J, Kauffmann A, van Dijk M, El-Kebir M, Visscher KM,
Holownia J, Macfarlane C, Bennion BJ, Gelpi-Dominguez S, Lightstone FC, van der Storm T, Geerke DP, Mark AE, Klau GW.
OFraMP:A Fragment-Based Tool to Facilitate the Parametrization of Large Molecules.
J. Comput. Aided Mol. Des., 2023, 37, 357-371. DOI:10.1007/s10822-023-00511-7
Validation
Stroet M, Caron B, Visscher K, Geerke D, Malde AK, Mark AE.
Automated Topology Builder version 3.0: Prediction of solvation free enthalpies in water and hexane.
J. Chem. Theory Comput. 2018, 14, 11, 5834-5845 DOI:10.1021/acs.jctc.8b00768
Koziara KB, Stroet M, Malde AK, Mark AE.
Testing and validation of the Automated Topology Builder (ATB) version 2.0: prediction of hydration free enthalpies.
J. Comput. Aided. Mol. Des., 2014, 28, 221-233. DOI:10.1007/s10822-014-9713-7
Related work
Zhou Z, Mark AE, Stroet M.
Engineering Transferable Atomic Force Fields: Empirical Optimization of Hydrocarbon Lennard-Jones Interactions by Direct Mapping of Parameter Space.
J. Chem. Theory Comput., 2023, 19, 4074-4087. DOI:10.1021/acs.jctc.3c00427
Kuschert S, Stroet M, Chin YKY, Conibear AC, Jia X, Lee T, Bartling CRO, Stromgaard K, Guntert P, Rosengren KJ, Mark AE, Mobli M.
Facilitating the structural characterisation of non-canonical amino acids in biomolecular NMR
Magnetic Resonance, 2023, 4, 57-72. DOI:doi.org/10.3929/ethz-b-000602558
Stroet M, Sanderson S, Sanzogni AV, Nada S, Lee T, Caron B, Mark AE, Burn PL.
PyThinFilm: Automated molecular dynamics simulation protocols for the generation of thin film morphologies
J. Chem. Inf. Model., 2023, 63, 2-8. DOI:doi.org/10.1021/acs.jcim.2c01334
Stroet M, Koziara KB, Malde AK, Mark AE.
Optimization of empirical force fields by parameter space mapping: A single-step perturbation approach.
J. Chem. Theory Comput., 2017, 13, 6201-6212. DOI:10.1021/acs.jctc.7b00800
Reisser S, Poger D, Stroet M, Mark AE.
Real cost of speed: The effect of a time-saving multiple-time-stepping algorithm on the accuracy of molecular dynamics simulations.
J. Chem. Theory Comput., 2017, 13, 2367-2372. DOI:10.1021/acs.jctc.7b00178
Malde AK, Stroet M, Caron B, Visscher K, Mark AE.
Predicting the prevalence of alternative warfarin tautomers in solution.
J. Chem. Theory Comput., 2018, 14, 4405-4415. DOI:10.1021/acs.jctc.8b00453
van Gunsteren WF, Daura X, Fuchs PFJ, Hansen N, Horta BAC, Hunenberger PH, Mark AE, Pechlaner M, Riniker S, Oostenbrink C
On the effect of the various assumptions and approximations used in molecular simulations on the properties of bio-molecular systems: Overview and perspective on issues
ChemPhysChem 2021, 22, 264-282. DOI:10.1002/cphc.202000968
Canzar S, El-Kebir M, Pool R, Elbassioni K, Malde AK, Mark AE, Geerke DP, Stougie L, Klau GW.
Charge group partitioning in biomolecular simulation.
J. Comput. Biol., 2013, 20, 188-198. DOI:10.1089/cmb.2012.0239
Engler MS, Caron B, Veen L, Geerke DP, Mark AE, Klau GW.
Multiple-choice knapsack for assigning partial atomic charges in drug-like molecules.
LIPIcs-Leibniz International Proceedings in Informatics 113, 2018, DOI:10.4230/LIPIcs.WABI.2018.16
Schmid N, Eichenberger AP, Choutko A, Riniker S, Winger M, Mark AE and van Gunsteren WF.
Definition and testing of the GROMOS force-field versions 54A7 and 54B7.
Eur. Biophys. J., 2011, 40, 843-856. DOI:10.1007/s00249-011-0700-9
Oostenbrink C, Villa A, Mark AE and van Gunsteren WF.
A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6.
J. Comput. Chem. 2004, 25, 1656-1676. DOI:10.1002/jcc.20090
The Automated Topology Builder (ATB) and Repository has been developed and is currently maintained with support from the University of Queensland (UQ), the Australian Research Council (ARC) and the Queensland Cyber Infrastructure Foundation (QCIF). Access to the ATB is provided free to academic users from publically funded teaching or research institutions. Access for academic use is conditional on: i) any molecule submitted to the ATB being made publically available and ii) the source of any material downloaded from the ATB being appropriately acknowledged in any publications or other forms in which research using this material is disseminated.
Use of the ATB by other parties, or academic users wishing to restrict the access of others to specific molecules, is considered to be commercial in nature. Commercial access is available by licence or collaborative agreement. Parties interested in commercial licencing or other arrangements should contact Prof Alan E. Mark at the address provided at the bottom of the page.
See the FAQ page to get started.
Tutorials for common tasks on the ATB are currently being developed, the first of which is available here: How to View an Existing Molecule
For queries regarding the ATB, please contact: 
This project is supported by the Australian Research Data Commons (ARDC). The ARDC is enabled by NCRIS.

